Frequently Asked Questions (FAQ)
FAQ 1. What are serial sections and why are they important?
Any of several microscopic sections made and arranged in consecutive order are called serial section. A continuous series of sections reveal structures in three dimensions (3D).
FAQ 2. What is step sectioning?
Step sectioning means taking a section and putting it on a slide, then shaving into the tissue a set distance, perhaps 100 microns or 500 microns, before taking the next section, and repeating this process. The result is a series of slides that show sections in “steps” or increments through the tissue.
FAQ 3. How do I prepare bone samples for tissue processing?
- fixation ( see “Routine FFPE Histology Services”, FAQ 1)
- After fixation, you can perform decalcification or you can send to VitroVivo for decalcification
- Method of decalcificationa:
a. Quick decalcification for H&E staing or histology special staining: Immerse tissue cassette in 11% formic acid with a stir bar overnight in a fume hood, Rinse in running water for 30- 60 minutes (the smell should be gone).
b. Slow EDTA decalcification for Immumostaining: Fixed specimens are rinsed in old EDTA decal solution before placing in Decal Bath. Solution must be stirred continuously. Decal time for mouse bone samples are as follows: Calvaria: 7 days; Arm or legs: 14 days.
FAQ 4. How do I make EDTA decalcification Solution?
Make Solution Recipe (4 Liter): Distilled Water 3 L; Hydroxide, concentrated 280 ml; EDTA (FW 292.2) 400 gm
Dilute 280 ml of concentrated Ammonium Hydroxide in 3L of distilled water. Add 400gm of EDTA and stir till dissolved. Add more concentrated ammonium hydroxide until pH is 7.2. Add more water to make the final volume 4L.
FAQ 5. How do I prepare 2D culture cell samples for making FFPE blocks?
- The volume of the packed cell pellet ideally needs to be approximately 0.5 mL. This requires approximately four 75 cm2 sized flasks, or two 150 cm2 flasks of near-confluent cell culture. Less material will result in a size-limited preparation, but this may be sufficient of only a few procedures are expected to be run.
- Scrape the cells: For adherent monolayer cells, do not trypsinize, as this may destroy cell-surface protein markers. Working quickly, pull the flasks from the incubator, and scrape the cells into the media. Transfer to a sterile 50 mL polypropylene centrifuge tube. Spin at room temperature for five minutes in swinging bucket centrifuge (setting 3 for 5 minutes in a standard clinical centrifuge, or approximately 200 x g).
- If using suspension cells, pellet 50 ml of cell culture supernatant in 50 ml conical tubes at 500xg for 10 min.
- Fixation: Aspirate media off cell pellet. Very slowly, add 20 ml neutral buffered formalin or zinc formalin (4º C) (contact the VitroVivo Biotech if you require this reagent), letting it flow gently down the side of the tube, in order not to disturb the pellet. You may re-centrifuge if the cell pellet is disturbed. The cells need to fix overnight at 4º C in formalin. You may bring the cell preparation the day of preparation, or after overnight incubation.
- If delivery of the preparation cannot be made within 24 hours of the start of fixation, remove the formalin and replace with 20 ml of 70% EtOH WITHOUT re-suspending the pellet. This will act as a non-crosslinking preservative, and the cells can be kept this way indefinitely at 4 º C. Do NOT freeze the cells.
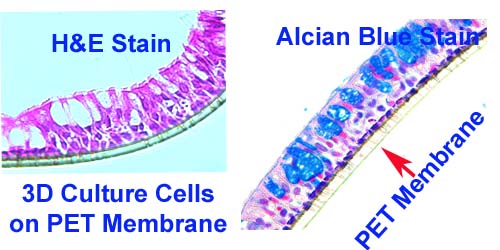
FAQ 6. How do I prepare 3D culture cell samples for making FFPE blocks?
- If the 3D culture cells are suspension sphere cells, the cell preparation is same as 2D culture cell (see FAQ3).
- If the 3 D culture cells grow on membrane ( such as PET membrane), cells should be fixed with 10% neutral buffered formalin for 30 min, then remove the formalin and replace with 70% EtOH.
- We will cut the membrane into 2-4 pieces and embed them in agarose gel for routine tissue processing.

Flow Chat: From 3D Culture Cells to Digital Images